

分野1レポート HPCによる計算生命科学
木寺 詔紀
Akinori Kidera
理化学研究所
HPCI計算生命科学推進プログラム
計算生命科学
計算生命科学と呼ばれる分野、特にそのHPCを基盤とした部分は、2006年に始まった「次世代生命体統合シミュレーションの研究開発」(2006-2012)というプロジェクトから出発したといえるだろう。実際には2006年のはるかに前から、生命の要素としての、生体分子を扱う分子シミュレーション、臓器、人体を扱う生体シミュレーション、生命情報を扱うバイオインフォマティクスなどは、すでにそれぞれ別のソサエティの中で長い歴史を刻んできていた。2006年は、それらの研究者すべてがはじめて一堂に会して、「生命体統合」という旗印を上げた年であった。ひとつのプロジェクトの中に集まることで、それぞれの分野が“異なった分解能・時間スケールで現れてくる様々な生命の階層”をどう捉えてきたかについての議論を積み重ねることで、ようやくそれらすべての階層を接続した全体像を構築するという共通の目標が見えてきた。そこにはじめてひとつの分野としての計算生命科学が誕生した。階層接続においては、上位階層と下位階層の時間・空間スケールのギャップを埋めるために、下位階層での長大な時間、巨大な空間の振る舞いを計算する必要が生じる。それこそが計算生命科学がHPCによる大規模計算を必要とする最大の理由である。そして2011年に、「京」を利用するHPCI戦略プログラム分野1「予測する生命科学・医療および創薬基盤」(2011-2016)において、本格的な階層接続の試みが開始された。
このように階層接続を目標とする計算生命科学は、生命科学自体のパラダイムシフトともいえる進展によって支えられている。近年の計測技術の著しい進歩の結果、様々な生命現象について、階層を越えた時間・空間の定量的な計測情報が大量に生み出されるようになってきた。それらのデータを統合して現象を動的なシステムとして統一的に説明するためのモデルが必要とされるなかで、HPCを用いた実験情報と整合的なシミュレーションモデルを構築する可能性が現れてきた。さらに、次世代シーケンサーのインパクトは、計算によって解析しなければならない生命ビッグデータという莫大な量の情報を生み出し、より多くの計算資源への要求、HPCへの期待につながっている。このように、生命科学が生命ビッグデータに基づいたデータ駆動型サイエンスに変貌しつつあるなかで、計算生命科学の役割はさらに重要性を増してきている。
生命科学のこのような状況は、計算生命科学の研究が、計測情報を与える実験現場、オミックスデータを与える医療現場との密接な連携のもと行われる必要性を増大させている。戦略プログラム分野1では、そのような連携体制をプロジェクトとして確立することをひとつのミッションとして掲げて活動を行ってきた。さらに、分野1のもうひとつの課題である創薬研究では、製薬企業との密接な連携のもと活動を行ってきた。
以下に、分野1の4課題についてその研究成果の代表例を説明する。
- 研究開発課題1:細胞内分子ダイナミクスのシミュレーション
この課題は、生命階層の最下層を扱う分子シミュレーションによる生体分子の研究分野である。階層接続の言葉を使えば、生体分子のレベルからその上層である細胞レベルへの接続を、計算規模を大幅に拡大することによって達成することが目的であるということができる。具体的には、細胞レベルに至る第一段階として、多数分子が細胞内で起こす細胞機能のシミュレーションを行っている。ここで取り上げる細胞機能としては、「細胞内信号伝達:細胞内の生体分子の物性、特にその拡散運動」と「転写制御:巨大な高次分子複合体の構造とダイナミクス」を取り上げた。
- 細胞環境を考慮した信号伝達経路のモデリング(理研 杉田有治)
細胞に外部からもたらされる信号・刺激を核にまで伝え応答反応をするために、様々な細胞内の生体分子間の相互作用が関与している。しかし、第0次近似的にいえば、生体分子が細胞質の中で拡散運動をすることで情報が伝わると考えることができる。ところが、細胞内は多数の生体分子が高い濃度で充填された状態にあり(分子混雑と呼ぶ)、その中での様々な低分子化合物を含む細胞環境下における生体高分子の相互作用と分子拡散がどのようなものであるかは分かっていない。そこで、マイコプラズマという最も小さな細菌の細胞質が含む生体分子によって構成される世界最大級の1億原子系の全原子分子動力学シミュレーションを実施した(図1;分子動力学計算プログラムGENESIS)。このシミュレーションによってはじめて、細胞環境での生体分子の状態、分子拡散を含めた原子レベルの細胞質ダイナミクスの実像を理解する道が拓かれた。
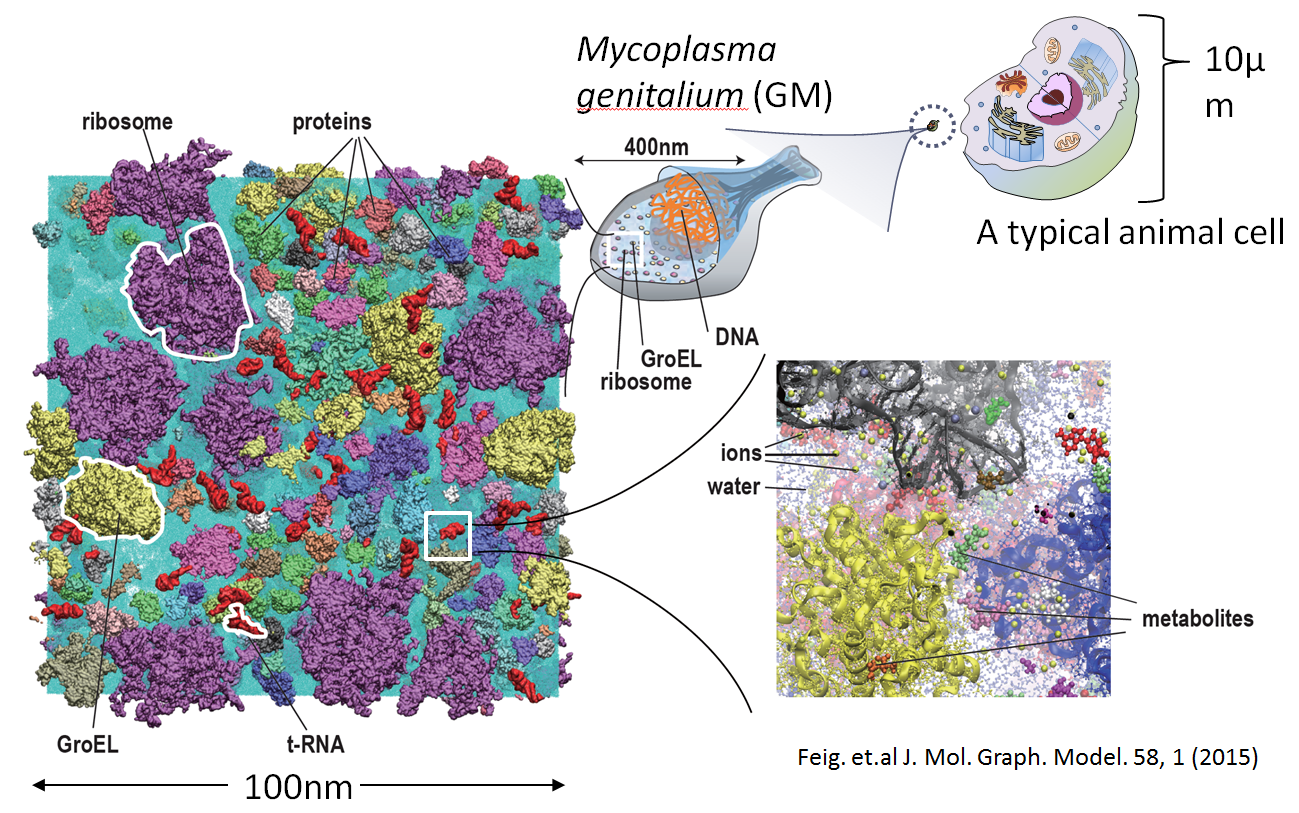
図1 マイコプラズマの細胞質部分の1億原子系の全原子分子動力学計算のスナップショット。マイコプラズマは、通常の動物細胞と比べるとはるかに小さい。そのさらに細胞質部分を1辺100nmの部分を切り出してきた。さらに拡大したところに示されているように、水中に多数のタンパク質が高度に充填され(細胞混雑)、そこにさらに大量の低分子化合物(代謝産物、イオン)が存在する(理研 杉田有治)
- ヌクレオソーム、クロマチンの機能発現機構(JAEA 河野秀俊、横浜市大 池口満徳、京大 高田彰二)
DNAから遺伝情報を読み取る過程である転写は、近年、クロマチンと呼ばれるDNAがヒストンタンパク質に巻き付いた巨大な高次複合体がダイナミックにその構造を変化させることによって、いつ、どの遺伝情報を転写されるかが制御されていることが分かってきた。そこで、クロマチンの構造とダイナミクスを明らかにするために、階層ごとに、粗視化分子モデル(粗視化モデル計算プログラムCafeMol)と全原子分子モデル(分子動力学計算プログラム SCUBA、Marble)とを用いた大規模マルチスケールシミュレーションを行っている。巨大でフレキシブルな構造を持つクロマチンのシミュレーション研究は、多くの生化学的実験、X線結晶構造解析、溶液X線小角散乱、電子顕微鏡などの多様な実験情報が前提となるところから、それらの実験研究との共同研究体制の中で実施され、計算、実験とそれぞれ相補的な情報を積み上げることで、転写制御という重要な細胞機能の解明に向けて研究を行っている。
- 細胞環境を考慮した信号伝達経路のモデリング(理研 杉田有治)
- 研究開発課題2:創薬応用シミュレーション(東大 藤谷秀章)
新規の医薬品を創り出す創薬研究は、実験、計算を含んだ極めて長い時間を要する探索研究である。その中で計算における最大のボトルネックは、標的タンパク質にどの候補化合物が十分な強度で結合するかどうかを評価する分子レベルの階層にある。一般には、計算量を節約するために経験的な評価関数が用いられているが、結合自由エネルギーにして5kcal/mol以上の誤差が避けられず、化合物の十分な絞り込みには至らず、実験による大量の結合アッセイ実験がどうしても避けられない。仮に、それらの化合物が化合物ライブラリの中に含まれる既存のものであれば、スクリーニングは容易であろうが、新規化合物の場合は、合成に著しく時間がかかり、創薬のプロセスを遅延させる理由のひとつとなってきた。そこで、1kcal/mol以下の結合自由エネルギーの精度を求めて、HPCを利用することによって分子シミュレーションの技術を用いた高精度な自由エネルギー計算を、多数の候補化合物について行い、創薬プロセスを加速することがこの課題の目的である。
計算は、標的タンパク質の薬剤候補化合物としてあらかじめデザインされた多数の新規化合物について、独自開発をしたMP-CAFEEという安定して高精度の自由エネルギー計算を実施し得るプロトコルによって行った。これは標準的な分子動力学プログラムGromacsに組み込まれ、国際的な評価を受けている。この計算方法を用いた場合、「京」を使う事によって、二、三ヶ月で数百個の薬剤候補化合物の結合自由エネルギー計算を高精度で行うことができる。
この課題で最も重要なことは、「京」による計算が、正しく創薬プロセスの中に組み込まれていることであり、それがまさにこの「京」を用いた課題において実現し、前臨床試験(動物を用いた薬物動態、薬効、安全性などについての試験)の段階に、ふたつの化合物医薬品、ひとつの抗体医薬品が到達する成功を収めた(図2)。
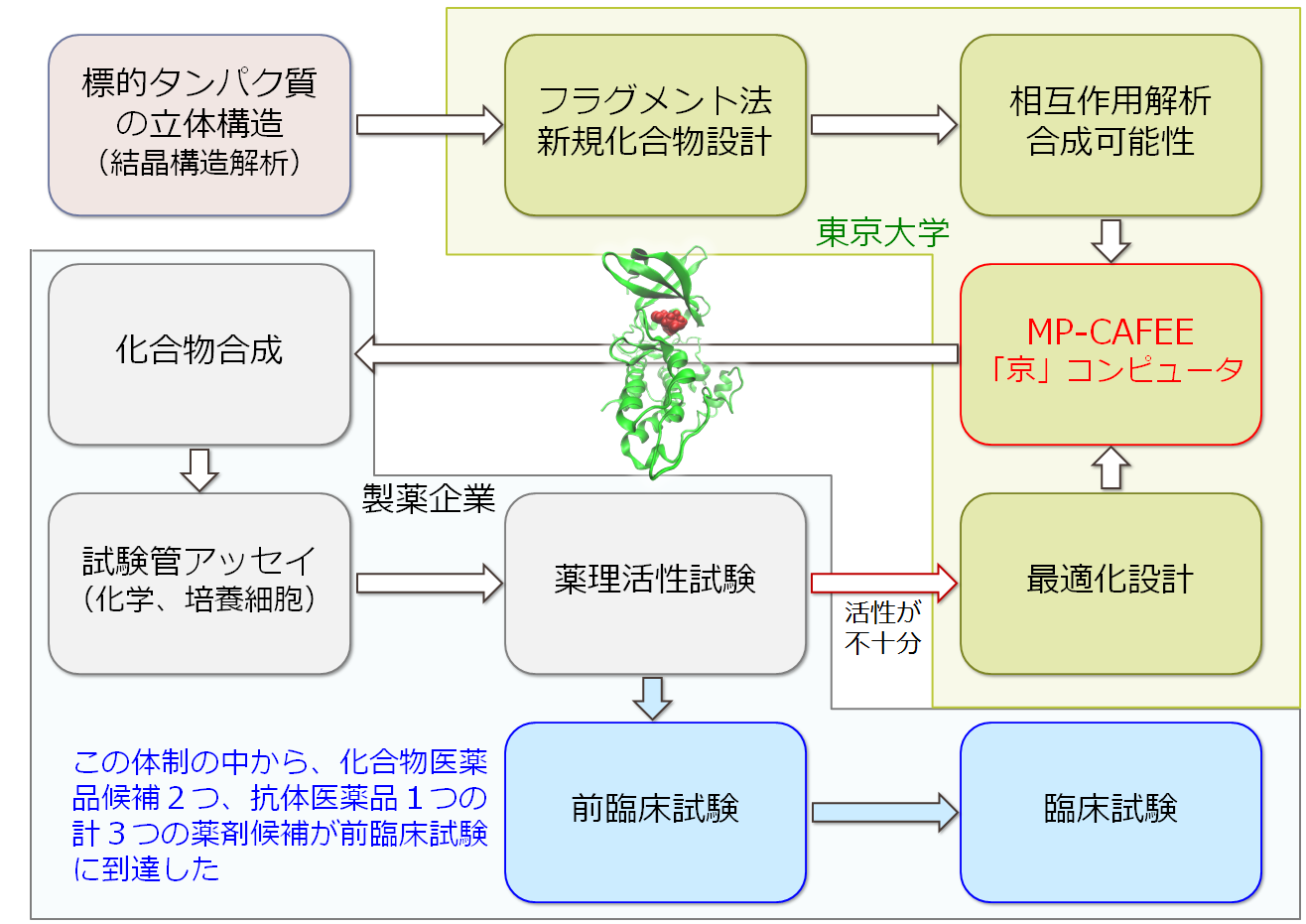
図2 課題2における製薬会社との共同研究体制。薬剤の標的タンパク質の結合評価の精度を「京」による結合自由エネルギー計算によって格段に向上させることで、合成すべき新規化合物の数を十分に減らすことが可能になり、創薬プロセスを著しく加速することが可能となった(東大 藤谷秀章)。
- 研究開発課題3:予測医療に向けた階層統合シミュレーション
この課題では、分子・細胞スケールから臓器・全身スケールに階層を接続することによって、分子レベルからの疾患モデルを構築し医療に貢献することを目指している。ここで、臓器としては、循環器系、脳神経系と連成した筋骨格系のふたつを選び、それらに関わる幅広い疾患に展開できる統合シミュレーターの構築を目指している。
- 心疾患の治療法・薬効評価のためのマルチスケール・マルチフィジックス心臓シミュレーション(東大・UT-Heart研 久田俊明、東大 高木周)
心臓シミュレータUT-Heartは、“筋収縮の最小単位である分子レベルのサルコメア”-“心筋細胞”-“心筋の繊維・層構造を反映した心臓”という3階層の統合を達成したシミュレータである(図3)。三層の階層接続は、サルコメアを構成するタンパク質の遺伝子に変異があることに起因する肥大型心筋症から、巨視的な心臓のかたちが先天的に正常とは異なる先天性心疾患までを対象として扱うことができる。それだけでなく、UT-Heartは、エネルギー代謝や電気生理学的状態をも再現する機能がある。さらに、この課題で開発してきた血栓シミュレータ(EX-THROM:血流中を流れる多数の赤血球や血小板などの血球細胞が、流れ場と相互作用して変形しながら流れていく中での、分子レベルの相互作用に基づく血小板凝集をシミュレートする)と接続することで、冠動脈の狭窄による狭心症、心筋梗塞のシミュレーションを行うことも視野に入れている。このように、UT-Heartは臨床現場から創薬現場まで幅広い応用の可能性を秘めた計算生命科学のキラーアプリということができるだろう。
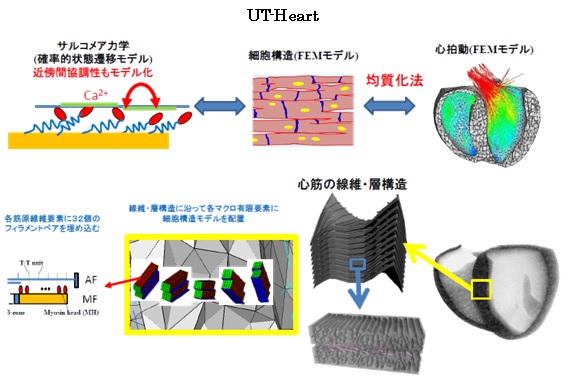
図3 心臓シミュレータUT-Heart。分子(モンテカルロ法)-細胞(有限要素法)-臓器(有限要素法)の3階層の統合。さらにその中に組み込まれている構造モデルを表示した(UT-Heart研 久田俊明)。
- 神経疾患による運動機能障害解明のための全身筋骨格-神経系統合シミュレーション(東大 高木周、OIST 銅谷賢治、東大 中村仁彦、阪大 野村泰伸)
脳神経系と連成した筋骨格系の研究では、パーキンソン病による運動障害のシミュレーションに挑戦している。パーキンソン病は、大脳基底核におけるドーパミンの不足により、手足のふるえ(振戦),筋固縮,動作緩慢,歩行障害などの運動障害を示す神経変性疾患の一つである。その病態の再現のために、世界最大級の細胞数の脳神経系シミュレーションに成功したNEST、筋線維の集合体として筋肉の振る舞いを再現するHi-MUSCLE、全身筋骨格シミュレータK-Bodyを、ソフトウェアMusicによって統合してシミュレーションを行った。ドーパミンの不足から生じる大脳基底核での約15ヘルツの振動を再現することに成功し、そのシグナルが視床で約半分の周波数になり、大脳皮質、脊髄から筋線維へと伝わり、パーキンソン病特有の手の震えを起こすことを見出した。脳神経系と筋骨格系という通常ではその関係をあらわに見ることのできないふたつの階層をシミュレーションの方法で統合することで、病態の正しい理解につなげることができるものと期待している。
- 心疾患の治療法・薬効評価のためのマルチスケール・マルチフィジックス心臓シミュレーション(東大・UT-Heart研 久田俊明、東大 高木周)
- 研究開発課題4:大規模生命データ解析
バイオインフォマティクスは、分子レベルの遺伝子の情報と、それが巨視的なレベルに発現する表現型というふたつの階層を接続することを目的とする分野である。このバイオインフォマティクスの課題では、次世代シーケンサーなどから得られる生命ビッグデータを解析することで、病態の分子レベルでの解明、薬剤標的タンパク質の同定、個人ゲノムに対応した医療への応用などをめざしている。
- 大規模データ解析によるがんのシステム異常の網羅的解析とその応用(東大 宮野悟)
がんはシステムの異常であるとされるように、単一の遺伝子ではなく、ゲノム全体に変異が起こり、生命システム全体に異常が現れる。その全体像を理解するためには、ゲノム配列の解析、遺伝子発現解析、エピゲノム(転写を制御する、遺伝子、ヒストンの化学修飾)解析などのがん細胞の広範な網羅的解析が必要となる。そして、そこから得られた膨大な情報をシステムとして記述し尽くさなければならない。図4は、乳児期にHTLV-1ウイルスに感染し、数十年後に発症し、発症後半年で死にいたる成人T細胞白血病リンパ腫(ATL)における遺伝子異常を、「京」でゲノム解析パイプラインGenomonを用いて解析した結果得られた全体像である。ATLは、これまで遺伝子異常の全体像が不明のままであったために治療成績が40年にわたって向上してきていなかったが、この大規模データ解析によってATLの全体像がようやく明らかにされ、今後の診断、治療法の進展につながるものと期待されている。この研究は、京都大学医学研究科 小川誠司教授との共同研究として行われた。
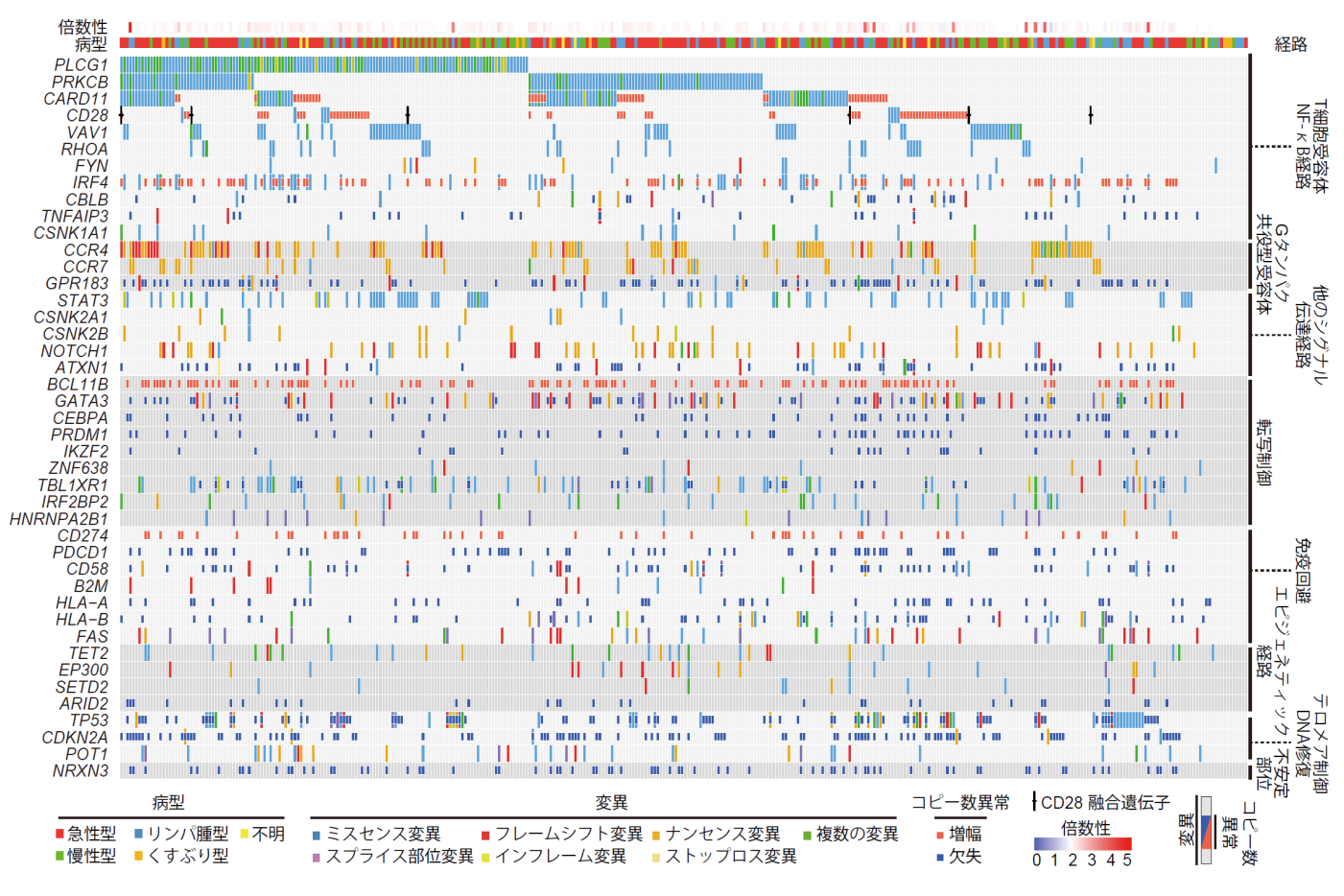
図4 成人T細胞白血病リンパ腫(ATL)における遺伝子異常の全体像。横軸は370のATLの症例を、縦軸には染色体の倍数性、病型、43個の顕著な変異を示す遺伝子(右欄外にはその遺伝子の関わる機能)を配置し、各遺伝子の変異、コピー数異常、CD28(上から4つ目の遺伝子)では融合遺伝子の解析結果を示している。ATLによってどの遺伝子にどのような異常が生じるかという全体像がひとつの図に集約されている(東大 宮野悟)。
- 大規模生体分子ネットワーク解析による脂肪細胞組織の刺激応答の網羅的解析とその応用(阪大 松田秀雄)
この研究は、遺伝子発現データから遺伝子ネットワーク(遺伝子間の因果関係をグラフとして表示したもの)を求めることで、まったくこれまでに想定されていなかった遺伝子の制御関係が発見されたものである。
白色脂肪細胞は脂肪を蓄積するが、その肥大化は肥満につながり、糖尿病など生活習慣病の大きなリスク要因となっている。近年、マウスの実験で一部の白色脂肪細胞が寒冷環境にさらされることによるアドレナリンの分泌で、ベージュ脂肪細胞という脂肪を熱として燃やす細胞に変化することが報告された。ヒト成人でもベージュ細胞の存在が確認され、肥満解消の観点から高い関心を集めている。しかし、なぜわずか一部の白色脂肪細胞のみしか変化しないという効率の悪さがあるかについては明らかではなかった。そこで、低温環境で飼育したマウスから取得した遺伝子発現データを用いて、約10,000個の遺伝子からなる遺伝子ネットワークをネットワーク解析ソフトBENIGNによって構築した。その結果、ベージュ脂肪細胞への変化を炎症に関連したIL-1βと呼ばれるタンパク質が抑制していることが分かった。このIL-1βとベージュ脂肪細胞への変化との関係はこれまでにまったく想定されていなかったものであり、「京」による大規模ネットワーク解析があってはじめて明らかになったものであると評価されている。この研究は、京都大学農学研究科 河田照雄 教授との共同研究で行われた。
- 大規模データ解析によるがんのシステム異常の網羅的解析とその応用(東大 宮野悟)
